中国科大团队在锂硫电池研究方面取得重要进展
- 时间:2018-09-12 01:15:09 来源:合肥微尺度物质科学国家研究中心|
锂硫(Li-S)电池因高理论比容量、能量密度以及低成本等优势而备受关注。但充放电过程中间产物多硫化锂的溶解引起穿梭效应,严重限制了其实际应用。为缓解该问题,目前主要通过添加各种碳材料及金属化合物来吸附多硫化锂。然而,在实际应用中,不同金属化合物在提升Li-S电池性能方面表现出巨大差异。如何从微观层面揭示Li-S电池性能提升的宏观现象背后的本质原因以及存在的科学规律,成为该领域极具挑战性的工作。
中国科学技术大学钱逸泰院士团队和王功名教授课题组通过实验和理论结合的方式,研究了金属钴基化合物(Co3O4, CoS2, Co4N以及CoP)在Li-S化学中的动力学行为,发现钴基化合物中阴离子的价电子的p能带中心相对费米能级的位置是影响Li-S电池界面电子转移反应动力学性质的主要因素。近日,该研究成果以《Deciphering the Modulation Essence of pBands in Co-based Compounds on Li-S Chemistry》为题,发表在Cell Press旗下的国际顶级能源材料期刊Joule杂志上。该论文第一作者是博士后周建斌,副研究员刘晓静和硕士研究生祝琳嵚。
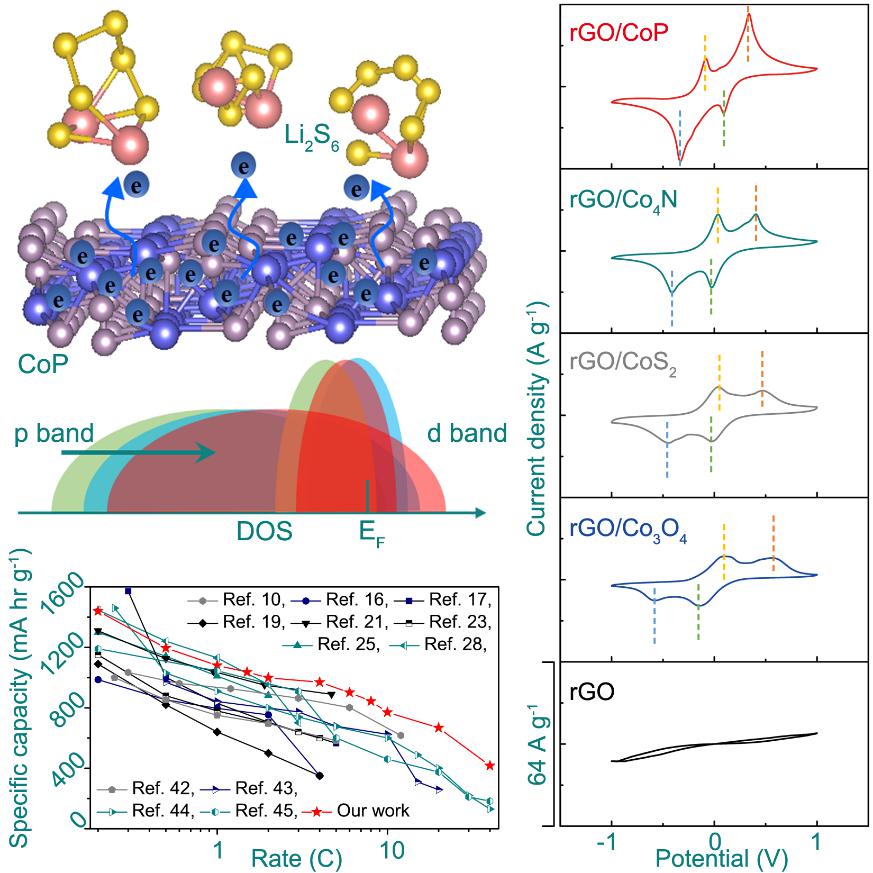
通过对rGO/Co-anions在Li2S6→Li2S的转化动力学性能的研究,发现制备的金属钴基化合物表现出完全不同的电化学动力学行为,其中rGO/CoP对多硫化合物转化的能垒最低,而且反应电流最大。随后,将rGO/Co-anions作为硫的载体,研究了一系列S@rGO/Co-anion复合材料在Li-S电池中的电化学性能。发现S@rGO/CoP正极材料的极化现象最小,而且倍率性能也最好的,甚至在40.0 C的条件下,容量仍有417.3 mA h g-1,对应的是当前最高的功率密度137.3 kW kg-1。
DFT模拟结果表明,相比于Co3O4, CoS2和Co4N,CoP对Li2S6以及Li2S的吸附能相对适中。同时电荷差分密度分析表明,吸附能与材料表面电荷的局域程度有很大的相关性;结合其电化学行为,发现过强或过弱的吸附能都不利于多硫化合物电化学转化的动力学性能,这与催化反应中的Sabatier理论一致;通过尝试关联不同钴基化合物的阴离子价带的p能带中心位置与多硫化合物电化学转化的动力学性能,发现改变阴离子价电子的p能带中心相对费米能级的位置,能够有效调控界面电子转移反应动力学,从而成为影响Li-S化学动力学性能的主要因素。
该项研究得到国家重大科学研究计划、国家自然科学基金等项目的资助。
论文链接:https://www.cell.com/joule/fulltext/S2542-4351(18)30395-7
中国科学技术大学钱逸泰院士团队和王功名教授课题组通过实验和理论结合的方式,研究了金属钴基化合物(Co3O4, CoS2, Co4N以及CoP)在Li-S化学中的动力学行为,发现钴基化合物中阴离子的价电子的p能带中心相对费米能级的位置是影响Li-S电池界面电子转移反应动力学性质的主要因素。近日,该研究成果以《Deciphering the Modulation Essence of pBands in Co-based Compounds on Li-S Chemistry》为题,发表在Cell Press旗下的国际顶级能源材料期刊Joule杂志上。该论文第一作者是博士后周建斌,副研究员刘晓静和硕士研究生祝琳嵚。
DFT模拟结果表明,相比于Co3O4, CoS2和Co4N,CoP对Li2S6以及Li2S的吸附能相对适中。同时电荷差分密度分析表明,吸附能与材料表面电荷的局域程度有很大的相关性;结合其电化学行为,发现过强或过弱的吸附能都不利于多硫化合物电化学转化的动力学性能,这与催化反应中的Sabatier理论一致;通过尝试关联不同钴基化合物的阴离子价带的p能带中心位置与多硫化合物电化学转化的动力学性能,发现改变阴离子价电子的p能带中心相对费米能级的位置,能够有效调控界面电子转移反应动力学,从而成为影响Li-S化学动力学性能的主要因素。
该项研究得到国家重大科学研究计划、国家自然科学基金等项目的资助。
论文链接:https://www.cell.com/joule/fulltext/S2542-4351(18)30395-7
相关文章
- 中国科大揭示记忆性ILC1s形成及维持机制2018-11-20 08:22:57
- 中国科大发现“老药”曲尼斯特可抑制NLRP3炎症小体并改善炎症性疾病2018-03-14 01:08:40
- 汪志勇教授研究组在GREEN CHEMISTRY上发表题为A metal-free decarboxylative cyclization ...的论文2011-03-28 02:11:00
- 汪志勇教授研究组在CHEMISTRY-A EUROPEAN JOURNAL上发表题为Highly Enantioselective Henry ...的论文2011-03-28 02:09:00
- 王均教授研究组在MOLECULAR PHARMACEUTICS上发表题为Targeted Delivery of Antisense...的论文2011-03-07 01:39:00
- 王均教授研究组与人合作在ACS NANO上发表题为Simultaneous Delivery of siRNA ...的论文2011-03-07 01:30:00